Protocols > Western blot This protocol covers protein preparation, NuPage (Novex) gel electrophoresis, blotting and antibody detection.
Important note: each protein-antibody pair may require specific optimization of this general protocol and this has to be taken into account. Proteins of low abundancy are best analyzed in specific cellular fractions. Caution should be taken when working with proteins which have high turnover or are light sensitive. If your band separation on a gel is not working well, analyze the sample composition buffers and conditions of your electrophoresis.
Protein isolation Total plant protein is isolated in 1x PEB buffer prepared freshly from 4x stock (Agrisera product no. AS08 300) according to the protocol given in the product info on the Agrisera website (fresh protease inhibitor cocktail must be included). For a protocol of protein isolation from diatoms, see: Protein isolation from diatoms.
Protein amounts are determined using 1/5 of the volumes of the standard DC Protein assay (BIORAD) against a BGG standard dilution curve of 0.0, 0.2, 0.4, 0.6 and 0.8 mg/ml (plus a 0.5 mg/ml reference) prepared freshly from 2.0 mg/ml stock (PIERCE) in 1x PEB. Absorbances are read in triplicates against distilled water. Variation of photometer readings should not exceed 5-10%, and r2 of the calculated standard curve should be > 0.995. Samples are stored aliquoted at -20ºC and used for no longer than 6 months.
[More information: product info sheet supplied from manufacturer for products used above] 
Sample preparation for loading
Protein extracts in 1x PEB are prepared for loading in equal amounts and equal volumes (0.5-10 µg in 10-15 µl) with 1x NuPage LDS Sample Buffer (Thermofisher/INVITROGEN) containing 50 mM DTT (added from freshly prepared 0.5 M stock within 30 min). Samples are incubated at 70ºC for 10 min, cooled down to RT, spun down shortly and loaded directly onto previously prepared gels. For each marker lane, a mixture of 2 µl MagicMark (Thermofisher/INVITROGEN) and 3 µl NovexSharp (Thermofisher/INVITROGEN) is prepared in the volume equivalent to the sample volume, containing 1x LDS sample buffer. The marker does not contain added DTT and is not heated prior to loading. When the aim of a Western blot is to detect plant membrane proteins, the samples should not be heated more than 70ºC for 5-10 min, and fresh 50 mM DTT should be used. 
Gel preparation prior to loading
Precasted 4-12% NuPage LDS-PAGE gels (Thermofisher/INVITROGEN) are prepared prior to loading as follows:
After unpacking comb, and lower slot strip are removed, the cassette is briefly rinsed in deionized water. All slots are individually spooled with 200 µl of 1x running buffer. Gels are inserted into the ShureLock Electrophoresis System (Thermofisher/INVITROGEN) and outer buffer chamber is filled with 1x running buffer (MES or MOPS). Following a check for leakage, the inner gel chamber is filled with 1x buffer containing NuPage Antioxidant (Thermofisher/INVITROGEN) according to the instructions of the manufacturer. 1x buffer is prepared freshly from 20x stock (Thermofisher/INVITROGEN) with deionized water at RT.
[More information: product info sheet from manufacturer for products used] 
LDS gel electrophoresis
Samples are loaded using a pipette with disposable tips (can also be loaded usin a Hamilton Syringe that is cleaned at least 3 times in 1x running buffer and passed over with a Kleenex tissue between loadings). Marker is loaded last. 
Electrophoretic separation is performed in the cold, in an ice bucket or in a cold room, to avoid distortion caused by excess heat. The applied voltage is 150 V until the front of the running buffer stain enters the lower cassette slot (approx. 60-70 min). Electrophoresis is stopped and cassettes are removed from the tank, opened by removing the shorter front plate. The gels are then either stained or taken for blotting. The voltage has to be adjusted accordingly, to avoid blasting the target protein through a membrane, depending on the pore size of the used membrane and the molecular weight of the target protein.
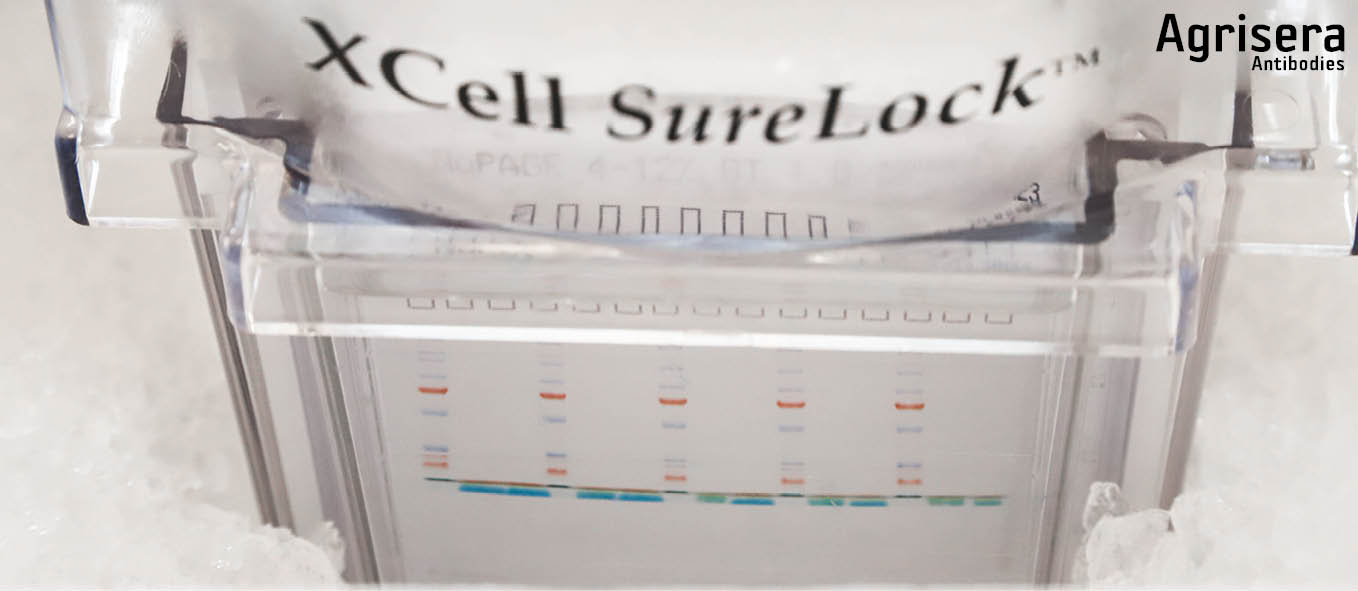
Blotting
All steps should be performed with clean gloves and forceps.
Prior to blotting of each gel, 1 membrane (nitrocellulose or PVDF) and 2 pieces of filter paper (WHATMAN) are cut exactly to the size of the NuPage gels (without stacking gels; 6.8 x 8.1 cm). 1x NuPage transfer buffer containing at least 5% (up to 20%) methanol is prepared from 20x stock (Thermofisher/INVITROGEN). The preparation of the blotting sandwich is as follows:
One filter paper is pre-wetted for some seconds in 1x NuPage transfer buffer and placed on the gel still attached to the longer back-plate of the gel-cassette. The cassette is flipped upside down and the gel is detached from the cassette with the tool supplied by the manufacturer (Thermofisher/INVITROGEN) with filter paper side down to a parafilm strip.
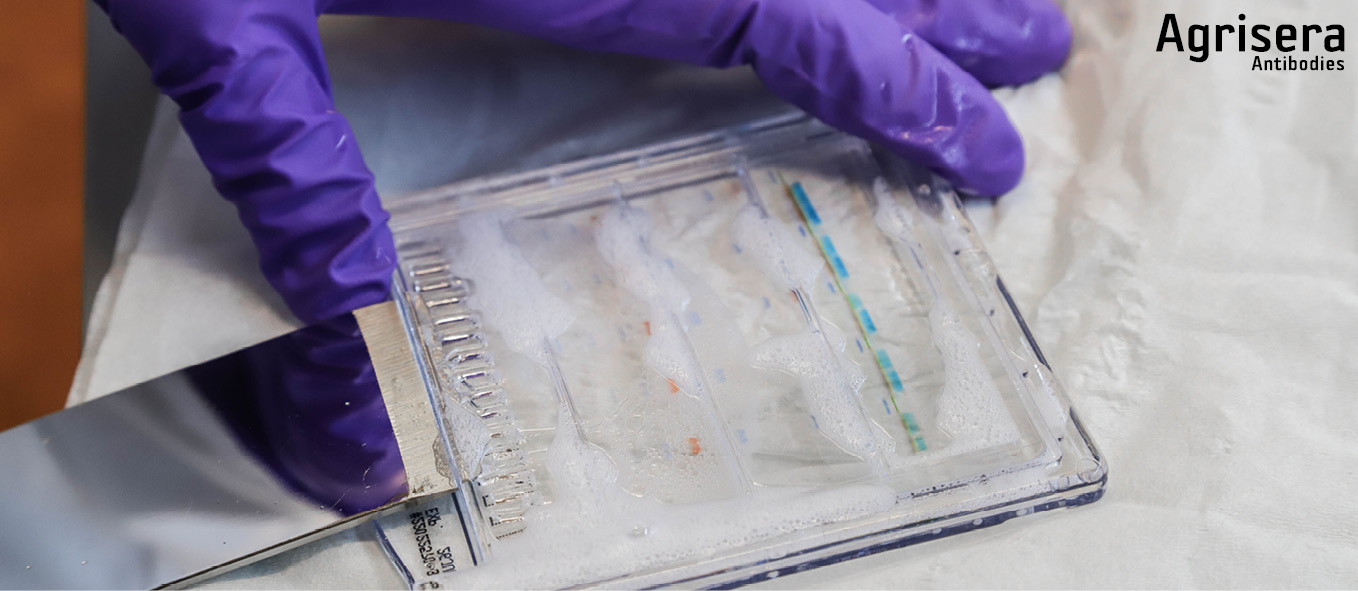
The PVDF membrane is pre-wetted for 1 min in 1x transfer buffer (for PVDF in addition pre-wet in 100% methanol for 1 min prior to that) and placed with exact fit onto the gel. The gel footer is cut off and the sandwich is completed with a filter paper (pre-wetted for some seconds in 1x NuPage transfer buffer, and placed with exact fit on topside of the membrane.

For each gel sandwich, 4-6 blotting pads are pre-wetted in ice cold 1x transfer buffer. 2-3 pads are placed in the cassette, the sandwich is positioned aligned to bottom of cassette, and covered with the 2-3 remaining pads. The pads should now exceed ~0.5 cm over the border of the cassette. By attaching the lock pads, the sandwich is pressed, and residual buffer is poured off. While firmly holding the cassette appressed, it is inserted in the tank and locked with the clamp. The cassette (1 cassette ~50 ml) is filled with 1x transfer buffer containing 1/1000 (v/v) NuPage antioxidant (Thermofisher/INVITROGEN), not higher than to the top of the blotting pads. The tank is firmly knocked down 8-10 times onto the table to remove air-bubbles. Transfer buffer is then added to the top of the blotting pads. The outer tank chamber is filled with ice cold deionized water and the tank is placed in the an bucket.
The blotting is performed at 30 V for 1-2 h, depending on the pore size of the used membrane, and the molecular weight of the target protein we aim to detect. The cassettes are then disassembled, and the membrane is washed in deionized water for 2-3 min (shaker, RT). Gels may subsequently be stained with Coomasie, to check transfer efficiency. The membrane is placed for drying, protein side up, between WHATMAN-paper. Membranes may be stored at RT in the dark for up to 6-12 months. Drying the membrane can help to keep proteins denatured, and throught this make specific epitopes be more accessible for antibodies.
Antibody incubation
All steps should be performed at RT with agitation.
The membrane is blocked 1 h with 5% low-fat milk powder in 1x TBS-T (20 mM Tris-Base, 137 mM NaCl, 0.1% TWEEN 20, pH 7.6). 1x TBS-T is freshly prepared from stocks of pH-adjusted 5x TBS (pH 7.6) and 10% (v/v) TWEEN 20. Blocking solution is discarded, and each membrane is incubated for 1 h with the desired dilution of primary antibody (1:500 to 1:50 000) in 10 ml of TBS-T containing 2% low-fat milk powder. Overnight incubation of primary antibodies might contribute to increased background signal, and should not be applied unless it is beneficial for obtaining a signal for some specific antibodies.
After discarding the primary antibody solution, the membrane is first washed briefly once (5 s) and then for 15 min followed by 3 additional washings of 5 min in 1x TBS-T (without blocking agent). The volume should be 10-20 ml per washing step, depending on the size of the membrane, and should be kept constant.
The membrane is incubated with the desired dilution of secondary antibody (1:10 000 to 1:100 000) in 10 ml of TBS-T containing 2% low-fat milk powder. After discarding the secondary antibody solution, the membrane is washed as described above and incubated in 1x TBS-T pending signal detection.
Signal detection
Chemiluminescent detection Detect the activity of HRP, coupled to the secondary antibody, using Agrisera ECL reagents:
AgriseraECL Bright (AS16 ECL-N) for low pico to mid femtogram detection (for proteins of medium and high expression), or the enhanced AgriseraECL SuperBright (AS16 ECL-S) for extreme low femtogram detection (for proteins of low expression).
The below instructions should be applied for both AgriseraECL Bright and AgriseraECL SuperBright:
Store reagents A and B in the darkness at 4-8°C. Mix equal volumes of reagent A and B (chemiluminescent substrate) in a clean container and equilibrate to room temperature 30 min before use. Prepare your membrane prior to the addition of chemiluminescent substrate, by washing it with the buffer used in your protocol. This will remove any background prior to substrate contact. Optimal visualization may be obtained up to 20 min after substrate contact. Usually, incubation for 2-5 min is optimal. Remove excess substrate by filter paper. Cover blot with clear plastic wrap or sheet protector, and expose either with x-ray film or CCD camera.
Chromogenic detection For laboratories which do not have a CCD camera, visualization of the target protein can be made using so-called chromogenic detection using Agrisera BCIP/NBT ALP Substrate (AS19 BCIP-NBT) or Agrisera BCIP/NBT Plus ALP Substrate solution (AS19 BCIP-NBT-PLUS). In such a case, the secondary antibody is conjugated to an enzyme called ALP (Alkaline phosphatase). The reaction is visualized directly on the membrane through color development, and the membrane can be stored in the lab book for a future reference. This detection system is not appropriate for proteins of low expression or to detect protein modifications, as phosphorylation.
The below instructions should be applied for both Agrisera BCIP/NBT ALP Substrate and Agrisera BCIP/NBT Plus ALP Substrate:
- Equilibrate reagent to room temperature before use.
- Following final binding reaction with an ALP labeled probe, wash the membrane or tissue sections in Tris-buffered saline or Tris/HCl containing 0.1% Tween 20. Do not use phosphate buffer for washing since this will inhibit ALP.
- Incubate the membrane in Agrisera Substrate solution, protected from light, for 5-15 minutes. Make sure the membrane is completely covered in solution. Depending on enzyme activity longer incubations may be necessary. If the concentration of ALP probe is too high, color might develop almost immediately, and the formazan deposit can flake of the membrane or tissue section. High concentration of ALP probe can also result in formazan deposits forming a thin line around the band or dot, with no deposit in the center. This requires further dilution of the ALP probe.
- Wash the membrane or tissue sections with deionized water to stop the enzyme reaction.
- Tissue sections can be counter stained in a 0.5% solution of neutral red that has been prepared in a 0.1 Mol L-1 acetate buffer, pH 5.2. Hematoxylin solutions will not provide sufficient contrast. A blue-purple precipitate will be localized at sites of ALP activity on the tissue sections.
- Dry membranes and store at room temperature protected from light. Purple bands or dots will be visible at the sites of ALP activity. In case of excessive background staining try increasing the number of washes of the washing time since this indicates an incomplete removal of non-bound ALP from the membrane.
Fluorescent detection
Fluorescent detection employs secondary antibody conjugated to a fluorescent dye and does not involve use of any detection reagents. The fluorescent signal is recorded by a CCD camera. The advantage of this method is that it allows multiplexing more than one antibody per assay, and detection of normalization and loading controls on the same blot. Agrisera offers a wide range of secondary antibodies, conjugated to fluorescent dye, which can be used for signal visualization.
The complete collection of Agrisera's secondary antibodies can be found here. |

